Computer
Simulations Show that the Hydrogen Radical Transfer Reaction Catalyzed by
Methylmalonyl-CoA Mutase and Coenzyme B12
is Dominated by Extreme Quantum Mechanical Tunneling
A subclass of the coenzyme B12-dependent isomerases catalyze chemically challenging carbon-skeleton rearrangements. For example, methylmalonyl-CoenzymeA mutase (MMCM) catalyzes the reversible isomerization of methylmalonyl to succinyl in both humans and bacteria. This reaction represents an intermediate step in the catabolism of the odd‑chain fatty acids, branched-chain amino acids and cholesterol, and its impairment results in methylmalonic aciduria.
Coenzyme B12 is an extensively modified porphyrin whose structure was determined by X-ray crystallography by Hodgkin in 1961.

The structure revealed, unexpectedly at the time, a cobalt-carbon bond to the deoxyadenosyl component of B12. Homolytic rupture of this bond is a key step in the catalyzed rearrangement, but atomistic details of the mechanism have remained elusive. Recent advances in computational enzyme kinetics have allowed for simulations that help to elucidate the mechanisms of enzyme-catalyzed reactions, and Agnieszka Dybala-Defratyka and Piotr Paneth of the Technical University of Lodz in Poland, Ruma Banerjee of the University of Nebraska, and Donald G. Truhlar of the University of Minnesota have now uncovered the atomistic details of the process and have shown that the experimental results, from the laboratory of Professor Banerjee, can be explained by extreme quantum mechanical tunneling. The results are published in the June 26 issue of the Proceedings of the National Academy of Sciences.
The bacterial MMCM studied in this work is a heterodimer (with units called α and β ) as shown in this figure:
|
|
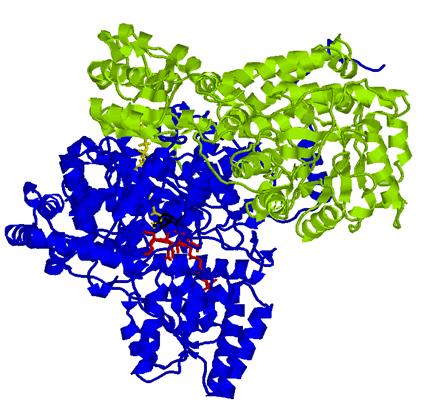
The α subunit (blue) contains the active site of the enzyme in which the cobalamin moiety of conenzyme B12 is in red, the substrate methylmalonyl-CoenzymeA is in yellow, and the 5´-deoxyadenosyl group of B12 is in black; the β subunit of the dimer is in green.
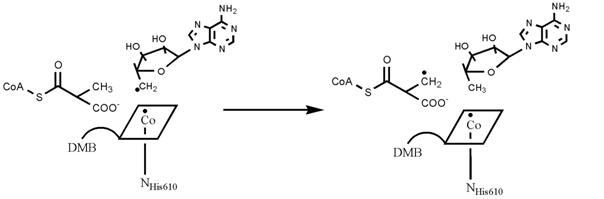
The key reaction is a hydrogen radical transfer as illustrated in the following scheme:
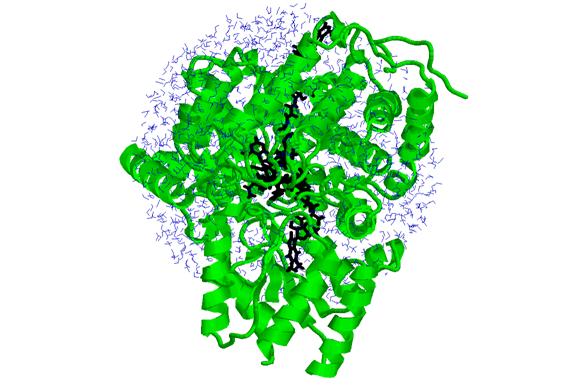
The atoms involved in the simulation are shown in the structure below, which contains 14878 atoms. This includes 672 amino acids (in green ribbons), 1388 water molecules (in blue), and 299 atoms of the active site residues (in black).
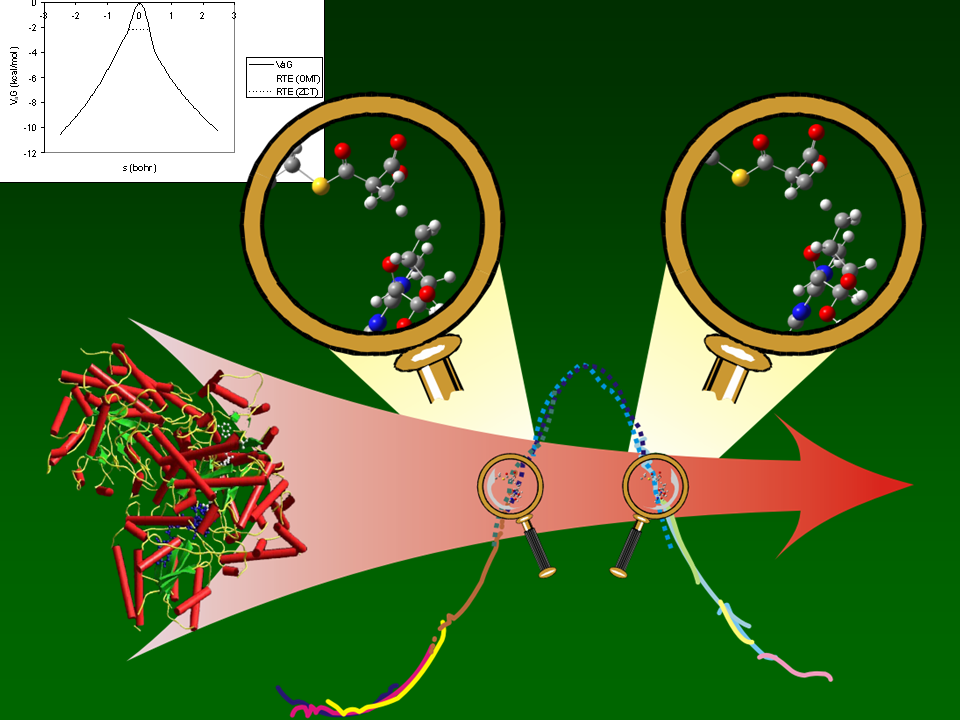
The simulations are based on transition state theory, which identifies the dynamical bottlenecks, called transition states, that represent points of no return along the reaction path. To a very good approximation, the rate of forming the dynamical bottleneck species is equal to the overall reaction rate. Attainment of the transition state may to likened to passing over a "mountain" pass or barrier; the bottleneck corresponds to the barrier top. In order to calculate quantitative reaction rate constants, eight transition state configurations were chosen, and an ensemble of eight reaction paths passing through these transition states was calculated. The total transmission coefficient, which is the factor by which tunneling increases the rate constant, was is obtained by averaging the dynamics over these eight paths.
The primary experimental observable is the kinetic isotope effect, which is the ratio of the reaction rate for the system shown in the scheme above to the reaction rate when the CH3 group that loses a hydrogen is replaced by CD3, where H denotes protium (the lightest isotope of hydrogen) and D denotes deuterium (a heavy isotope of hydrogen). Protium atom is lighter than deuterium and has a larger quantum mechanical zero point energy and a larger probability of tunneling. Both of these differences contribute to the reaction rate being higher for H than for D. In fact the experimental kinetic isotope effect is very dramatic—it is 49. That is, the rate constant for the CH3 case is 49 times larger for hydrogen transfer than for deuterium transfer. However, the calculations show that in the absence of tunneling, the kinetic isotope effect would be only 14. When tunneling is included the calculated kinetic isotope effect is increased to 51, in excellent agreement with experiment. This provide confidence in the detailed dynamic picture of the reactive event that is afforded by the computer simulation.
Tunneling corresponds to the system passing through the "mountain" rather than over the "mountain" pass. This could not occur for macroscopic objects, but because the H and D are very light they are described by the laws of quantum mechanics rather than the laws that govern the behavior of macroscopic objects. The simulation shows that the H reaction is speeded up by a factor of 93 due to tunneling, and the D reaction is speeded up by a factor of 26. A factor of 93 means that only 1% of the reaction proceeds in the classical way, by passing over the barrier. When tunneling dominates a reaction to this extent, it is called extreme tunneling.
Because the final results of the quantum mechanical atomistic simulation agree with experiment so well, the researchers were able to analyze them to better understand the nature of the tunneling events. They found that the tunneling of H or D is strongly coupled to motion of the other atoms in the active site of the enzyme, and they were able to identify the geometrical configuration at the critical configuration of the tunneling process. It is very gratifying that 46 years after the structure of coenzyme B12 was determined, the atomistic details of this biologically important reaction that it catalyzes have now been elucidated.
This work was supported in part by Polish State Committee for Scientific Research, the Fogarty International Research Collaboration Award, the National Institutes of Health, the National Science Foundation, and the University of Minnesota Supercomputing Institute.