|
|
Comp Chem Research Developments | |
| Archive of Comp Chem Research News | |
| February 1st, 2006 | |
| Simulating Fluid Phase Equilibria of Water from First Principles | |
|
Water holds a unique role among liquids, not only because of its ubiquity and importance on Earth, but more so because of its anomalous liquid properties. Water's ability to act as donor and acceptor for two hydrogen bonds, which leads to the formation of a tetrahedral network, and its participation in many chemical processes, particularly self-dissociation and acid-base equilibria, have made understanding its properties a grand challenge for liquid state theory and molecular simulation. The phase diagram holds a central role in thermodynamics and only with its knowledge it is possible to make meaningful comparisons of experiment, theory and simulation. For example, it would not be helpful to discuss whether a given water model can reproduce the well-known liquid-phase anomalies, such as the density maximum at T = 277 K and p = 1 atm or the (isothermal) compressibility minimum at T = 320 K and p = 1 atm, without first demonstrating that the liquid phase is actually thermodynamically stable at these conditions for the specific model. Graduate student Matthew McGrath, Professor Ilja Siepmann and a team of international collaborators are the first to successfully to compute from first principles the vapor-liquid coexistence curve of water. To this extent, efficient Monte Carlo algorithms were coupled with a mixed-basis set electronic structure program. This advanced simulation program, called CP2K, is freely available. A water representation based on the Becke-Lee-Yang-Parr exchange and correlation functionals yields a saturated liquid density of 0.9 g/ml at 323 K, and normal boiling and critical temperatures of 350 and 550 K, respectively. An analysis of the structural and electronic properties of the saturated liquid phase shows an increase of the asymmetry of the local hydrogen-bonded structure despite the persistence of a four-fold coordination, and decreases of the molecular dipole moment and of the spread of the lowest unoccupied molecular orbital with increasing temperature. Full details of this work are published in the Truhlar Festschrift. [McGrath, et al., J. Phys. Chem. A 110, 640-646 (2006)]
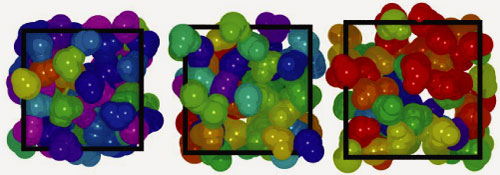
Snapshots of the saturated liquid phase of BLYP-GTH-TZV2P-1200 water at T = 323, 423, and 523 K (left to right). The molecules are colored according to their instantaneous molecular dipole moment, µ, ranging from red (µ < 1.9 D) to purple (µ < 3.2 D) in intervals of 0.1 D. |
|
|
|
|
|
The development of advanced computational strategies for the most challenging problems in chemistry and chemical physics is a theme common to the research endeavors of the Minnesota Computational Chemistry Group, where research includes new theoretical formulations, the development of new computational algorithms, and use of state-of-the-art supercomputers to solve prototype problems to high accuracy and to predict chemically useful results for a wide range of system scales ranging from a few atoms to thousands of atoms. Financial support from the National Science Foundation, Divisions of Chemical and Transport Systems and of Analytical and Surface Chemistry, is gratefully acknowledged. Part of the computer resources were provided by the Minnesota Supercomputing Institute. |
|
|
| |