

|
|
Comp Chem Research Developments | |
| Archive of Comp Chem Research News | |
| February 15th, 2006 | |
| Critical Properties of Aluminum | |
|
Aluminum is one of the most ubiquitous elements on Earth. Since its isolation in 1825, aluminum has found many technological uses but its physical properties are well known only at low reduced temperatures. Due to their strong cohesive interactions, the critical properties of most metals are not amenable to conventional experiments. Various extrapolation methods have been used to estimate the critical temperature of aluminum from low-temperature experimental data, but these estimates scatter over a range from 5700 to 12100 K. The poor precision of these estimates impedes technological advances and is an embarassment for our field. A team consisting of postdoctoral associates Divesh Bhatt and Ahren Jasper, graduate student Nate Schultz, and Professors Ilja Siepmann and Don Truhlar performed Gibbs ensemble Monte Carlo calculations using an embedded-atom potential to obtain the vapor-liquid coexistence curve and the critical properties for elemental aluminum. The embedded-atom potential, named NP-B, was parametrized to reproduce accurate energies for clusters and nanoparticles obtained from density functional theory. The calculations show that this potential yields accurate liquid densities, heats of vaporization, and saturated vapor pressures at low temperature where experimental data are available. The critical temperature for aluminum predicted using this potential is close to 6300 K, near the lower end of the range of values (5700-12100 K) extrapolated from experimental data. This work shpws how advances in computational materials science can answer fundamental questions about the properties of the elements that have remained too difficult for direct experimental measurement even 180 years after the isolation of aluminum. Full details of this work will be published in a March 2006 issue of JACS.
|
|
|
|
|
|
The development of advanced computational strategies for the most challenging problems in chemistry and chemical physics is a theme common to the research endeavors of the Minnesota Computational Chemistry Group, where research includes new theoretical formulations, the development of new computational algorithms, and use of state-of-the-art supercomputers to solve prototype problems to high accuracy and to predict chemically useful results for a wide range of system scales ranging from a few atoms to thousands of atoms. Financial support from the National Science Foundation and the Defense-University Research Initiative in Nanotechnology (DURINT) is gratefully acknowledged. Part of the computer resources were provided by the Minnesota Supercomputing Institute. |
|
|
| |
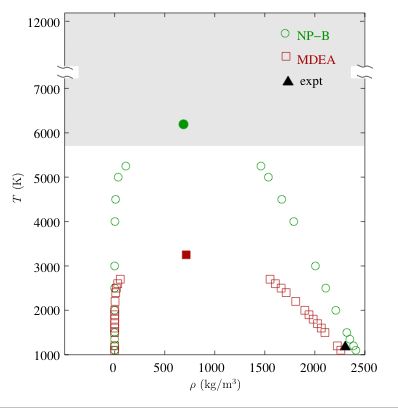 Vapor–liquid
coexistence curves of Al calculated using the
MDEA and NP-B potentials. The MDEA embedded-atom
potential was fitted by Mei and Davenport using only the cohesive energy
and bulk modulus of solid aluminum. The open and filled symbols denote
the saturated densities and the estimated critical points, respectively.
The shaded area represents the range of critical temperatures estimated
from experimental data and the filled triangle shows the experimental
liquid density of molten Al at 1173 K.
Vapor–liquid
coexistence curves of Al calculated using the
MDEA and NP-B potentials. The MDEA embedded-atom
potential was fitted by Mei and Davenport using only the cohesive energy
and bulk modulus of solid aluminum. The open and filled symbols denote
the saturated densities and the estimated critical points, respectively.
The shaded area represents the range of critical temperatures estimated
from experimental data and the filled triangle shows the experimental
liquid density of molten Al at 1173 K.