|
|
Comp Chem Research Developments | |
| Archive of Comp Chem Research News | |
|
November 24, 2004 | ||||
| ||||
|
The study of biochemical reactions using combined quantum
mechanical/molecular mechanical (QM/MM) methods has gained tremendous
attention. Biological reactions occur in a complex solvated
macromolecular environment where electrostatic effects play are often
attributed as being a main source of catalytic activity. The
challenge in theoretical studies of biochemical mechanisms is to move
accurate quantum electronic structure calculations from the gas phase into
complex biological environments. A key step toward this end involves
the accurate and efficient modeling of electrostatic interactions for
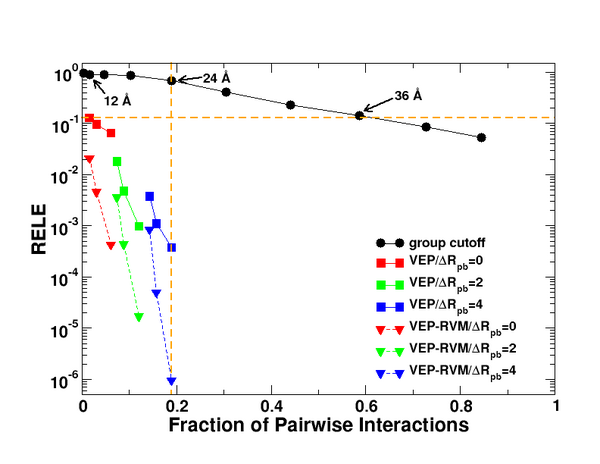
these very large systems. Recently, the research group of Prof. Darrin York of the Department of Chemistry has
made several advances in the design of new methods to treat long range
electrostatic interactions in QM/MM simulations of biochemical
reactions. Prof. Darrin York, along with graduate student Brent
Gregersen have recently introduced a new method for treatment of
long-ranged electrostatic interactions in QM/MM stochastic boundary
molecular dynamics (SBMD) simulations. The method allows the
electrostatic environment due to a tremendously large system to be
accurately modeled in an enzyme's active site for a fraction of the
computational cost of direct methods (Figure 1). The first
publication on the Variational Electrostatic Projection (VEP) method is
currently in press in the Journal of Physical
Chemistry B.
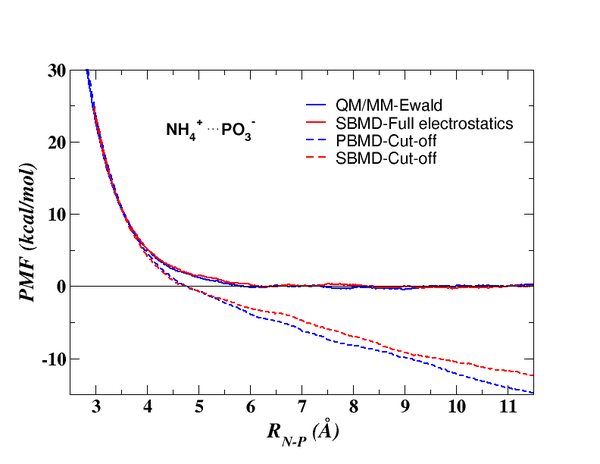
In some instances, it is important to apply more rigorous full periodic boundary simulations with explicit solvent. Although for molecular mechanical force field models, linear-scaling electrostatic methods for periodic systems exist, these methods have been sluggish in being generalized to combined QM/MM potentials due to the added complexity of the quantum mechanical electron density. Recently, Professor York, along with graduate student Kwangho Nam and Prof. Jiali Gao have developed a linear-scaling Ewald method for combined QM/MM simulations. Application of the linear-scaling QM/MM-Ewald method (Figure 2) demonstrates the importance of rigorous treatment of electrostatic interactions of reactions, especially those that involve ionic transition states or intermediates. The linear-scaling QM/MM-Ewald method is in press and is scheduled to appear in the first issue of the new ACS journal, Journal of Chemical Theory and Computation, in January, 2005.
| ||||
|
| ||||