

|
|
Comp Chem Research Developments | |
| Archive of Comp Chem Research News | |
| June 23rd, 2004 | |
| Liquid Water from First Principles: Validation of Different Sampling Approaches | |
|
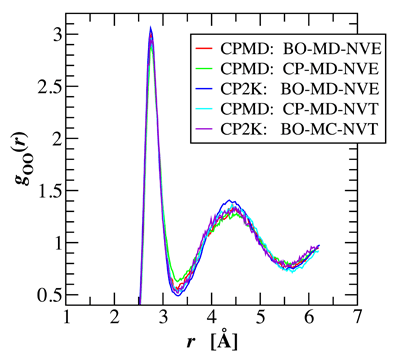
Water holds a unique role among liquids, not only because of its ubiquity and importance on earth, but more so because of its anomalous liquid properties. Thus, understanding its properties has been a grand challenge for liquid state theory and molecular simulation. The first particle-based simulations of liquid water using pairwise empirical potentials were carried out almost 40 years ago [1]. However, the strong dipole moment and large polarizability of water and its participation in many chemical processes, particularly its self-dissociation, pose a challenge for empirical potentials. Although great strides have been made in the development of empirical force fields for water, none of these has yet succeeded to yield a quantitative description of the thermodynamic, structural, and dynamic properties of water over its entire liquid range [2]. In contrast, an ab initio representation of water affords the opportunity to study both physical and chemical properties of water Graduate student Matthew McGrath, Professor Ilja Siepmann and a team of international collaborators have carried out a series of first principles molecular dynamics and Monte Carlo simulations for liquid water to assess the validity and reproducibility of different sampling approaches [3]. These simulations include Car-Parrinello molecular dynamics simulations using the program CPMD with different values of the fictitious electron mass in the microcanonical and canonical ensembles, Born-Oppenheimer molecular dynamics using the programs CPMD and CP2K in the microcanonical ensemble, and Metropolis Monte Carlo using CP2K in the canonical ensemble. It is found that the structural and thermodynamic properties of these simulations are in good agreement with each other as long as adiabatic sampling is maintained in the Car-Parrinello molecular dynamics simulations either by choosing a sufficiently small fictitious mass in the microcanonical ensemble or by Nose-Hoover thermostats in the canonical ensemble. Using the Becke-Lee-Yang-Parr exchange and correlation energy functionals and norm-conserving pseudopotentials, simulations at a fixed density of 1.0 g/mL and a temperature close to 315 K yield a height of the first peak in the oxygen-oxygen radial distribution function of about 3.0 (see figure) and a classical constant-volume heat capacity of about 70 J/mol K. | |
|
| |
| |
| |
|
The development of advanced computational strategies for the most challenging problems in chemistry and chemical physics is a theme common to the research endeavors of the Minnesota Computational Chemistry Group, where research includes new theoretical formulations, the development of new computational algorithms, and use of state-of-the-art supercomputers to solve prototype problems to high accuracy and to predict chemically useful results for a wide range of system scales ranging from a few atoms to thousands of atoms. Financial support from the National Science Foundation and the Department of Energy is gratefully acknowledged.[1] Barker, J. P.; Watts, R. O. Chem. Phys. Lett. 1969, 3, 144. [2] Chen, B.; Potoff, J. J.; Siepmann, J. I. J. Phys. Chem. B 2000, 104, 2378. [3] I.-F.W. Kuo, C.J. Mundy, M.J. McGrath, J.I. Siepmann, J. VandeVondele, M. Sprik, J. Hutter, B. Chen, M.L. Klein, F. Mohamed, M. Krack, and M. Parrinello, J. Phys. Chem. B, submitted for publication. | |
|
| |