|
|
Comp Chem Research Developments | |
| Archive of Comp Chem Research News | |
| January 21th, 2004 | |
|
| |
|
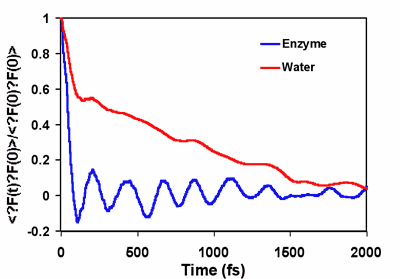
The origin of the rate acceleration achieved by enzymes is one of the fundamental questions in molecular biology. In principle, the rate enhancement in enzyme reactions can be achieved by lowering the free energy of activation and/or by increasing the generalized transmission coefficient in comparison with an equivalent uncatalyzed reaction in water [1]. Experimental and computational studies show that the dominant factor responsible for the rate acceleration is the reduction of the activation barrier in the enzyme, which is an equilibrium, thermodynamic effect and can be attained both by transition state stabilization and by reactant state destabilization through interactions with specific residues. The role of dynamics on enzyme catalysis, which is mainly due to the difference in the generalized transmission coefficient between the catalyzed and uncatalyzed reaction [1], remains to be fully understood. Reported in the Journal of the American Chemical Society (JACS, 2004, 126, ASAP), graduate student Kwangho Nam, Lakshmi S. Devi-Kesavan and Xavier Prat-Resina, and postdoctoral associate Mireia Garcia-Viloca in Professor Jiali Gao's group carried out reactive flux molecular dynamics simulations to study the dynamics of the nucleophilic substitution reaction of dichloroethane by a carboxylate group in haloalkane dehalogenase and in water. The classical reflection contribution (recrossing), i.e., the k(T) factor, to catalysis was determined using a combined QM/MM potential. Molecular dynamics simulations reveal that protein dynamics accelerates the reaction rate by a factor of 2 over the uncatalyzed reaction, a substantial effect in computation of the reaction rate, but the protein dynamic contribution to the overall rate acceleration by the enzyme is relative small compared to barrier reduction, which is about 11 kcal/mol. Most significantly, analyses of the friction kernel reveal that the origins of the reaction dynamics in water and in the enzyme are different. In aqueous solution, there is significant electrostatic solvation effect, which is reflected by the slow reorganization relaxation of the solvent. On the other hand, there is no strong electrostatic coupling in the enzyme and the major effect on reaction coordinate motion is intramolecular energy relaxation. [1] Garcia-Viloca, M.; Gao, J.; Karplus, M.; Truhlar, D. G. Science 2004, 303, 186-195. | |
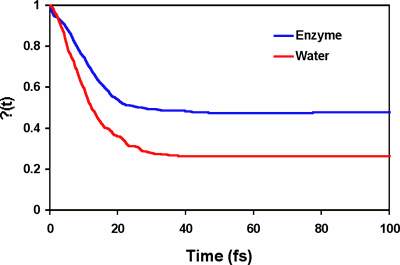
Computed time-dependent transmission coefficient, k(t), for the reaction in water (red) and in haloalkane dehalogenase (blue).
Autocorrelation function <dF(t) dF(0)> where dF(t) is the fluctuation in the force on the reaction coordinate at the transition state. | |
|
| |