|
|
Comp Chem Research Developments | |
| Archive of Comp Chem Research News | |
| April 14th, 2004 | |||||
|
| |||||
|
The modeling of dispersion forces is of considerable
interest in chemical physics and biomolecular simulations.
However, accurate quantum mechanical calculation of dispersion forces is
extremely challenging owing to the large basis sets and high degree of
electron correlation required for their description. Hartree-Fock
and conventional density-functional quantum models have traditionally
been unsatisfactory, and higher-level ab initio methods such as high-order
perturbation theory or coupled cluster approaches can only be applied to
very small systems due to large computational requirements. Recently, graduate student Timothy Giese and Prof. Darrin York of the
Department of Chemistry have designed a new quantum method for accurate
determination of dispersion interactions. The method is know
as a multi-coefficient correlation method for van der Waals (MCCM-vdW)
interactions that utilizes the transferability of basis set and electron
correlation effects to derive a model that captures dispersion effects
at a fraction of the computational cost of other comparably accurate
quantum methods. The method does not require use of so-called
"counterpoise corrections", and agrees extremely closely with both
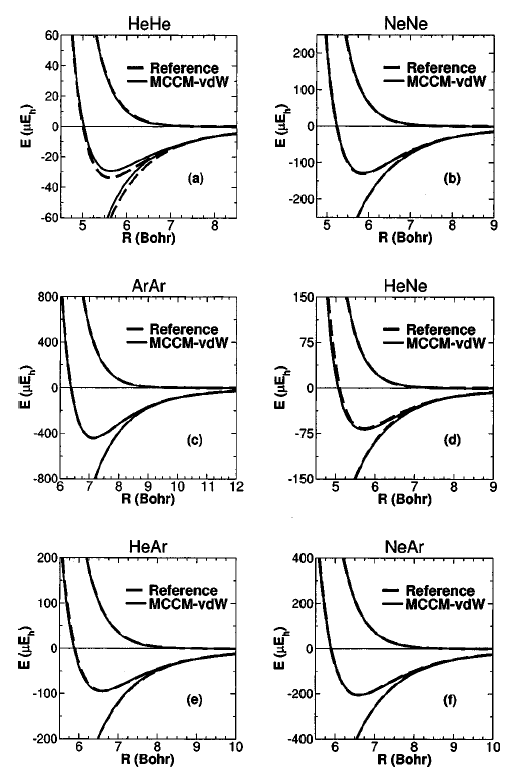
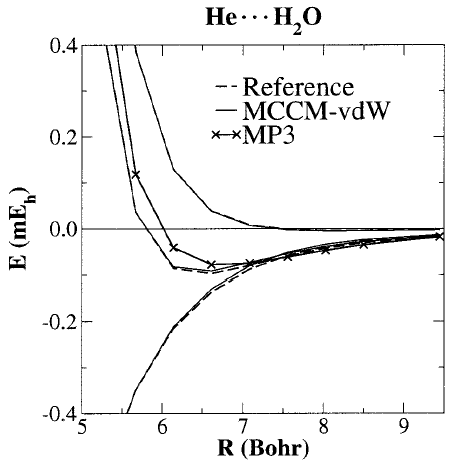
experiment and high-level quantum results (Fig. 1).
| |||||
|
| |||||
| The method can be used for determination
of potential energy surfaces such as rare-gas probes used to derive
dispersion potentials for molecular simulation force fields (often
performed at the MP2 or MP3 levels that are considerably less accurate),
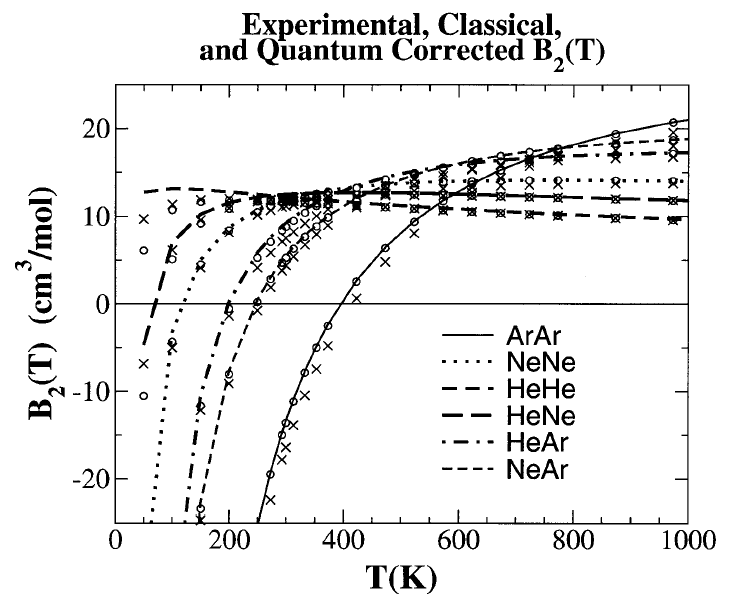
properties such as second virial coefficients (Fig. 2), and many-body
interaction potentials. The method opens the door toward the
reliable calculation of dispersion interactions of larger systems that may
provide benchmark data used to design new, extremely fast and accurate
semi-empirical quantum models for hybrid quantum mechanical/molecular
mechanical simulations of biological reactions. | |||||
|
| |||||