

|
|
Comp Chem Research Developments | |
| Archive of Comp Chem Research News | |
| July 9, 2003 | |
|
| |
|
Temperature is one of the most often used parameters to control solution processes, including protein folding or DNA/RNA denaturation, liquid-liquid extraction, and chromatographic separations. Despite its ubiquitous use, however, there is incomplete understanding of the factors that govern the effect of temperature on solution free energies (Gibbs free energies of transfer) and a common assumption is that the temperature pre-factor in the entropic term dominates the overall temperature dependence. Accordingly, a linear fit in a ``van't Hoff plot'' is often employed to extract an enthalpy that is viewed to be constant over the temperature range of interest. Graduate student Collin Wick, Professor Ilja Siepmann and Mark Schure (Rohm and Haas) have used configurational-bias Monte Carlo simulations in the Gibbs ensemble to calculate precisely the Gibbs free energies, enthalpies, and entropies of transfer for water, 1-butanol, and n-octane between their own liquid phase and a helium vapor phase at equilibrium. The simulation results (see Figure below) demonstrate that the temperature dependence of the Gibbs free energy of transfer is mainly driven by the variation of the enthalpy of transfer, that is the heat capacities of transfer. In contrast, the changes in the temperature-entropy term are small because the increase in the temperature is compensated by a decrease in the entropy of transfer. The decrease in the magnitudes of the enthalpy and entropy of transfer with increasing temperature can be explained by the decrease in cohesive energy density of the liquid phase (lower density at higher temperature) and a reduction of the entropic penalty for cavity formation and solvent re-organization (more free volume at higher temperature). | |
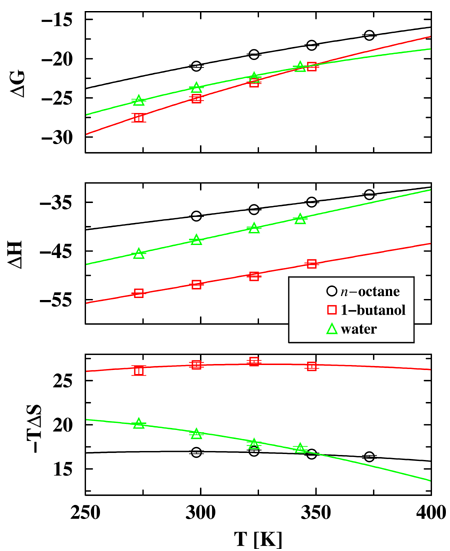
| |
|
Figure: Gibbs free energies, enthalpies, and temperature-entropy values (all given in units of kJ/mol) as functions of temperature. The lines show least-square fits using the constant-pressure heat capacities of transfer determined from the enthalpy or entropy derivatives. | |
|
The development of advanced computational strategies for the most challenging problems in chemistry and chemical physics is a theme common to the research endeavors of the Minnesota Computational Chemistry Group, where research includes new theoretical formulations, the development of new computational algorithms, and use of state-of-the-art supercomputers to solve prototype problems to high accuracy and to predict chemically useful results for a wide range of system scales ranging from a few atoms to thousands of atoms. Financial support from the National Science Foundation, Divisions of Chemical and Transport Systems and of Analytical and Surface Chemistry, and a Department of Energy Computational Science Graduate Fellowship is gratefully acknowledged. Part of the computer resources were provided by the Minnesota Supercomputing Institute. | |
|
|