|
|
Comp Chem Research Developments | |
| Archive of Comp Chem Research News | |
|
Jan 8, 2003 | |
| |
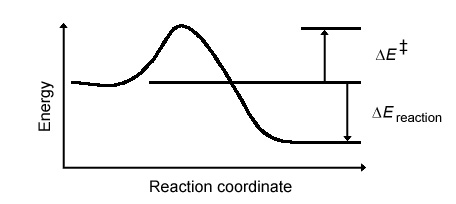
New methods by graduate student Benjamin J. Lynch and Professor Donald G. Truhlar are extending the realm of high-accuracy quantum chemical calculations up to larger molecules than previously affordable. The new methods provide tools that are well suited for the prediction of accurate chemical reaction rates. Computational thermochemistry and computational thermochemical kinetics are based on the Born-Oppenheimer approximation and the use of quantum-mechanical electronic structure theory to calculate potential energy surfaces. The electronic structure methods may be based on interacting-electron wave functions or on density functional theory. To quantify the performance of modern electronic structure methods, a database of atomization energies, energies of reaction (DEreaction), reaction barrier heights (DE‡), ionization potentials, and electron affinities was developed. The values of these chemical properties come experimental data, although extracting barrier heights from experiment is indirect, coming from a combination of well validated dynamical theory and experimental reaction rates.
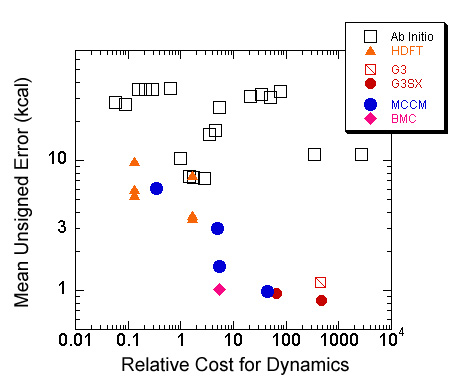
Explicitly correlated ab initio methods require very expensive calculations to attain reasonable accuracy for kinetics calculations. Hybrid density functional theory (HDFT) provides a very cost effective route for generating potential energy surfaces, and specialized HDFT methods such as MPW1K have been developed in the Truhlar group. However, HDFT methods have variable accuracy and are not systematicalyy improvable, and so the group has sought methods that are more accurate than HDFT, but still have a reasonable cost. Multi coefficient correlation methods (MCCM) are a class of methods first developed in the Truhlar group which calculate the energy using a linear combination of explcitly correlated calculations to extrapolate to the exact result. Many MCCM methods have been developed in the Truhlar group (MC-QCISD/3, MCG3/3) and the method has also become adopted by John Pople, Larry Curtiss, and coworkers (G3S, G3SX, G3SX(MP3)). The mean unsigned error for the best of these methods is ~1 kcal/mol for data in the database. For bond energies this corresponds to ~0.2 kcal/mol per bond The most recent progress has been in the developement of a balanced multicoefficient method (BMC). This method uses a new one-electron basis set to more accurately extrapolate to the correct answer. The BMC method has a slightly lower RMS error than G3SX(MP3) and is a factor of 12 lower in cost. | |
 |
|
|
Figure Caption Error (kcal/mol) vs. Cost for many popular electronic structure methods and for the new MCCM and BMC methods. | |
|
| |