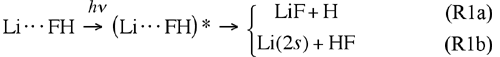|
|
Comp Chem Research Developments | |
| Archive of Comp Chem Research News | |
|
Oct 16, 2002 |
|
|
|
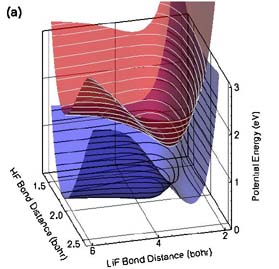
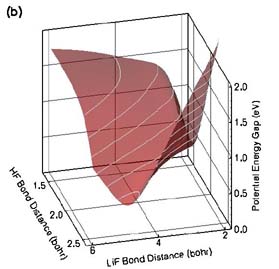
Calculations by Ahren W. Jasper, Michael D. Hack, Arindam Chakraborty, and Donald G. Truhlar are providing new insight into the transition states for reactions of electronically excited species. The Born-Oppenheimer (BO) approximation recognizes that electrons move orders of magnitude faster than nuclei, and therefore nuclear motion is effectively governed by its interaction with the average field created by the electrons. This insight allows one first to solve for the electronic motion (which may be represented in the form of a potential energy surface or PES), and then use the PES to determine the nuclear motion. Chemical systems that can be accurately modeled in this way are called BO systems or adiabatic chemical systems. There is an important class of chemical reactions for which the BO approximation is not valid, and these non-BO or nonadiabatic chemical systems require special consideration. Typical non-BO processes include chemical reactions of electronically excited species, ultraviolet photodissociation, nonadiabatic charge-transfer, chemiluminescence, etc. For these systems, more than one electronic state is important in the overall dynamics, and more than one PES needs to be included when solving for the nuclear motion. Furthermore, the nuclear motions corresponding to each of the PESs are coupled to one another, and therefore the nuclear motion for all of the PESs must be solved simultaneously. The LiFH system is a prototype non-BO system, and we have recently developed a set of coupled PESs for the LiFH system shown in part (a) of the figure. The ground-state potential energy surface (blue with black contours) has a van der Waals well in the Li(2s) + HF entrance valley and a barrier in the LiF + H exit valley. A strongly bound excited-state complex (exciplex) is present in the first excited-state (red with white contours) at a geometry similar to (but tighter than) the geometry of the ground-state van der Waals well. The ground and first-excited states of the LiFH system are coupled nonadiabatically, forming a seam of avoided crossing at larger Li–F and H–F separations, as evidenced by the small gap in part (b) of the figure. A new natural decay of mixing algorithm has been developed to provide an improved treatment of the decay of coherence in wave packets representing electronically nonadiabatic processes. This method has been used to study the process:
where an asterisk denotes electronic excitation. In this process, the van der Waals molecule is laser excited and can either produce ground-state Li or react to form LiF, and it is important to understand how to tune the laser to control the branching ratio. The calculations on the new set of coupled PESs show that the lifetime of the LiFH exciplex is shorter and less dependent on excitation wavelength than the lifetime of the NaFH exciplex. |
|
 |  |
|
Figure Caption (a) Coupled potential energy surfaces for the non-BO LiFH system, shown for linear geometries. The excited electronic state is shown red with white contours, and the ground electronic state is shown blue with black contours. Electronically excited Li(2p) interacts with HF to form a quasibound excited-state complex called an exciplex. Nuclear motion in the exciplex is strongly coupled to the ground electronic state, especially at large HF separations. This coupling induces electronic transitions, causing the exciplex to decay to form LiF + H and Li(2s) + HF. (b) Energy gap between the two potential energy surfaces. Note the small energy gap (~0.9 eV) towards large HF separations. |
|
|
| |