

|
|
Comp Chem Research Developments | |
| Archive of Comp Chem Research News | |
|
Dec 11, 2002 |
|
|
|
Understanding binary nucleation is a first step toward the goal of explaining multi-component nucleation that plays a critical role in many processes of atmospheric, environmental, and technological importance. In particular, binary mixtures consisting of water/alcohol or water/alkane have been the focus of many experimental investigations. Using sophisticated techniques, these experiments have provided data on the dependencies of binary nucleation rates on temperature, vapor pressure, and vapor composition. An important conclusion has emerged from the experimental studies, namely that the classical nucleation theory (CNT), which is based on the so-called capillary approximation and has been satisfactorily applied to numerous one-component systems, is incapable of describing the nucleation behavior for many binary systems. For some non-ideal mixtures, such as water/alcohol systems, CNT can even produce unphysical results. The deficiency of CNT arises from the fact that it fails to predict the differences in composition between the critical nucleus and the bulk liquid. This deficiency can lead to negative aggregation numbers for the surface active species and a prediction of decreasing nucleation rates with increasing partial pressure in violation of the nucleation theorem. Graduate student Bin Chen (now postdoc at the University of Pennsylvania) and Professor Ilja Siepmann of the Department of Chemistry have used a combination of the aggregation-volume-bias and configurational-bias Monte Carlo algorithms and the umbrella sampling technique to investigate two different binary vapor-liquid nucleation systems: water/ethanol and water/n-nonane. The simulations are able to reproduce the different non-ideal nucleation behavior observed experimentally for these two systems, i.e., the mutual enhancement of nucleation rates for water/ethanol mixtures and the two-pathway nucleation for water/n-nonane mixtures. Structural analysis provides microscopic explanations for the observed nucleation behavior. In particular, the simulations show a large and size-dependent surface enrichment of ethanol in the water/ethanol droplets, which confirms the previous experimental interpretation for this system. The immiscibility observed even for small water/n-nonane clusters causes the two-pathway nucleation mechanism. A detailed description of this work has been accepted for publication in The Journal of the American Chemical Society. |
|
 |
 |
 |
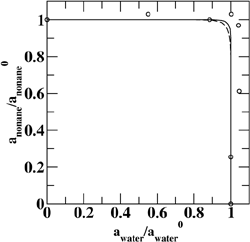 |
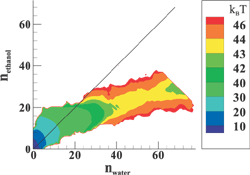
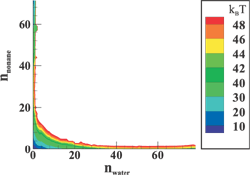
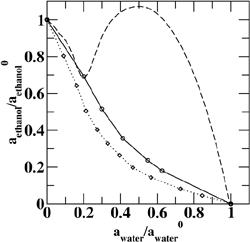
(Top left) Contours of the two-dimensional nucleation free energy surface as a function of the number of water and ethanol molecules calculated for an equimolar gas mixture. (Top right) Contours of the two-dimensional nucleation free energy surface as a function of the number of water and n-nonane molecules calculated for a nonane-rich gas mixture. (Bottom left) Comparison of the reduced onset activities for binary nucleation of water/ethanol mixtures at 260~K: experimental data (dotted line and diamonds), CNT predictions (dashed line) [Viisanen, Y.; Strey, R.; Laaksonen, A.; Kulmala, M. J. Chem. Phys. 1994, 100, 6062-6072], and simulation results (solid line and circles). (Bottom right) Comparison of the reduced onset activities for binary nucleation of water/n-nonane mixtures at 230~K: experimental data (circles), predictions using the nucleation theorem (dashed line) [Wagner, P. E.; Strey, R. J. Phys. Chem. B 2001, 105, 11656-11661], and the simulations results (solid line). |
|
The development of advanced computational strategies for the most challenging problems in chemistry and chemical physics is a theme common to the research endeavors of the Minnesota Computational Chemistry Group, where research includes new theoretical formulations, the development of new computational algorithms, and use of state-of-the-art supercomputers to solve prototype problems to high accuracy and to predict chemically useful results for a wide range of system scales ranging from a few atoms to thousands of atoms. Financial support from the National Science Foundation, Divisions of Chemical and Transport Systems and of Analytical and Surface Chemistry, is gratefully acknowledged. Part of the computer resources were provided by the Minnesota Supercomputing Institute. |
|
|
| |