

|
|
Comp Chem Research Developments | |
| Archive of Comp Chem Research News | |
November 1, 2000
|
|
| |
|
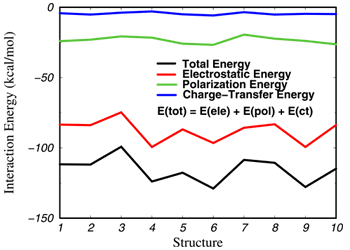
Hydrogen-bonding interaction plays an essential role in chemistry and biology. A fundamental question in understanding hydrogen-bonding interactions is the relative contribution of individual energy components to the total binding energy. State-of-the-art molecular mechanics force fields for computer modeling of protein and DNA systems utilize a simple electrostatic model to describe hydrogen-bonding interaction, which ignores the polarization and charge transfer effects. Consequently, a major effort in developing the next-generation force field for biological simulations is to incorporate explicit many-body polarization and perhaps charge transfer terms into these computations. The difficulty, however, is that little is known about the quantitative contribution of polarization and charge transfer effects in hydrogen bonding interactions. In a paper published in the Journal of Chemical Physics, 112 (2000), 5530, postdoctoral research associate Dr. Yirong Mo and Professor Jiali Gao, in collaboration with Professor Sigrid D. Peyerimhoff of Universität Bonn, Germany, developed a novel energy decomposition method using a block-localized wave function technique, which allows specific energy components in hydrogen-boding complexes to be determined. This energy decomposition method was shown to yield consistent and stable results as the size of the basis set in ab initio quantum mechanical calculations increases, a major improvement over previous methods. Applying this energy decomposition technique, Dr. Yirong Mo and NSF-Lando summer undergraduate student Heidi Privett investigated the electrostatic, polarization and charge-transfer contribution in a variety of hydrogen-bonded complexes in the gas phase and in aqueous solution. To elucidate the significance of polarization effects in describing hydrogen-bonding interactions, energy decomposition calculations were performed on computers provided by the Minnesota Supercomputing Institute for a series of selected configurations from combined quantum mechanical and molecular mechanical (QM/MM) Monte Carlo simulations. In this study, the solute and water molecules that are within 5 Å from any solute atom (about 23-27 water molecules) were treated by ab initio HF/6-311+G(d,p) theory, which are embedded in a box of ca. 500 water molecules represented by the TIP3P model. The figure shows a histogram of the energy decomposition results for 10 individual structures for an acetate anion in water (selected at every 100,000 configurations during the MC simulation). On average, the total interaction energy of CH3CO2- in water is -115.5 kcal/mol, of which -87.6 kcal/mol (76%) are due to electrostatic interactions, -23.3 kcal/mol (20%) are from polarization effects, and -4.5 kcal/mol (4%) originate from charge transfer. Natural population analysis revealed that only about 0.02 electrons migrate from the acetate anion into the aqueous solution. Similar trends were also found for other solute molecules representing functional groups of biological interest. These results suggest that improvement of molecular mechanics force fields may be focused on incorporation of the polarization effect. |
|
|
|
|
| |