

|
|
Comp Chem Research Developments | |
| Archive of Comp Chem Research News | |
November 15, 2000
|
|
| |
|
There is tremendous recent progress in using quantum mechanics for the accurate simulation of chemical processes. For example, a recent (July 26, 2000) NetStep news item discussed the simulation of quantum effects on hydride transfer reactions in an enzyme system with over 5000 atoms. There are many interesting problems for much smaller systems too. For example, HF dimer is the simplest neutral molecule with a hydrogen bond, and there has been considerable interest in the ab initio calculation of its vibrational-rotational energy levels to spectroscopic accuracy. This represents a prototype four-body problem. Progress in quantum mechanics, like classical mechanics, has often been marked by well known benchmarks on three-body systems, which are still challenging in many branches of physical chemistry and chemical physics. Four-body problems are usually much harder. Very recently, in a paper scheduled for publication in Chemical Physics Letters, Professor Donald G. Truhlar and coworkers Yuri Volobuev and Bill Necochea (both former graduate students in the Chemical Physics Program and Chemistry Department) have reported a calculation on the HF dimer ground-state energy that is converged to better about 0.1 part per million, which is probably the most accurate quantum mechanical calculation on a four-body system that has ever been carried out. The calculation was made possible by combining an original computational strategy for basis set selection with an original quadrature scheme designed for this problem and with a second-generation strategy for parallel computation. The largest calculation involved diagonalizing a dense matrix of order 29,541 whose formation required the calculation of 436,320,570 nine-dimensional integrals, but the whole calculation required only 5 hours and 24 minutes of computation on 32 processors of the University’s IBM SP supercomputer. The development of advanced computational strategies for the most recalcitrant problems in chemical physics is a theme running through many of the projects in the department's Computational Chemistry Group, where research includes new theoretical formulations, the development of new computational strategies and algorithms, and use of state-of-the-art supercomputers to solve prototype problems to high accuracy and to predict chemically useful results for a wide range of system scales ranging from a few atoms to thousands of atoms. |
|
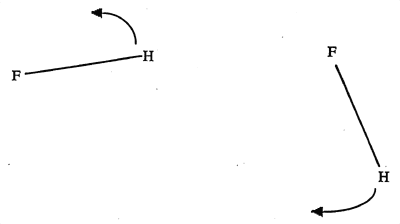 The HF dimer can execute an asymmetric stretch vibration in which the donor hydrogen becomes the acceptor, and the acceptor becomes the donor. The final state is quantum mechanically indistinguishable form the initial one; hence this is called a degenerate rearrangement, and it leads to a tunneling splitting of all the vibrational levels, similar to the splitting upon which the ammonia maser is based. |
|
|
| |