

|
|
Comp Chem Research Developments | |
| Archive of Comp Chem Research News | |
November 29, 2000
|
|
| |
|
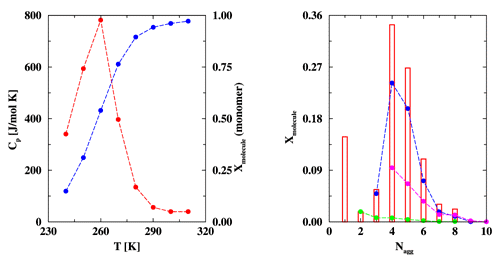
Strongly associating fluids are an important class of systems featuring intermolecular attractions with deep (a few tens of kJ/mol or thousands of Kelvin in thermal units), but narrow wells usually caused by the formation of intermolecular hydrogen bonds. These systems exhibit large deviations from the ideal-gas or ideal-solution behavior due to the formation of aggregates. Strongly associating fluids pose a special challenge for molecular simulation because aggregate formation leads to traps in phase space, i.e., configurations that have a very large Boltzmann weight associated with the large favorable energies of cluster formation. However, these "bonded" configurations represent only a small fraction of the total phase space compared to the large amount of non-bonded configurations. Graduate student Bin Chen and Professor Ilja Siepmann of the Department of Chemistry have developed the novel aggregation-volume-bias Monte Carlo (AVBMC) algorithm which greatly increases the sampling efficiency for strongly associating fluids. The AVBMC algorithm makes use of an unsymmetric Markov matrix to increase the transition probability for moves that lead to the formation of bonded configurations, and to enhance the acceptance rate for moves that lead to the destruction of bonded configurations. Using the AVBMC algorithm the properties of superheated hydrogen fluoride (HF) vapor were investigated. A plot of the constant-pressure heat capacity versus temperature (see Figure) shows an unusual peak for HF vapor that is reminiscent of a phase transition. The microscopic origin of this peak is a "transition" from an aggregate state at low temperatures to a monomeric state at high temperature, i.e. the concentration of monomeric (non-associated) molecules changes over a small temperature range (see Figure). Molecular simulation can also provide detailed information on the properties of the aggregates at a microscopic-level that would be difficult to measure experimentally. From a cluster type distrubtion (see Figure) we can learn that tetramers and pentamers are the dominant clusters at low temperatures and that most of the larger clusters have branched or cyclic structures. | |
 | |
|
(Left:) Constant-pressure heat capacity (red) and mole fraction of
monomeric molecules (blue) versus temperature for the superheated
HF vapor at a pressure of 15.5 kPa. The development of advanced computational strategies for the most challenging problems in chemistry and chemical physics is a theme common to the research endeavors of the Minnesota Computational Chemistry Group, where research includes new theoretical formulations, the development of new computational algorithms, and use of state-of-the-art supercomputers to solve prototype problems to high accuracy and to predict chemically useful results for a wide range of system scales ranging from a few atoms to thousands of atoms. A brief description of this research work can be found in The Journal of Physical Chemistry B 2000, 104, 8725-8734. Financial support from the National Science Foundation, Divisions of Chemical and Transport Systems, is gratefully acknowledged. Bin Chen would like to thank the Graduate School, University of Minnesota, for the award of a Stanwood Johnston Memorial Fellowship and a Doctoral Dissertation Fellowship. Part of the computer resources were provided by the Minnesota Supercomputing Institute. |
|
|
| |